Library Screens
Library based, high throughput drug design
Many biologic therapeutic modalities are easy to synthesize in combinatorial libraries, including antibodies, RNA binding therapies, viral vectors, and more. Using these methods with high-throughput library screening, biotechs are able to evaluate the fitness of hundreds of thousands of potential drug candidates against different targets or other types of selection.

Manage NGS experiments
Biotechs often use sequencing to count performant library members after an enrichment assay, resulting in large FASTQ files. These files are difficult to share, move around, and explore.
Here a bioinformatician populates Latch Data. They may upload files from AWS s3, BaseSpace, FTP, SRA, or local computers. They may do this from the comfort of their terminal. is a cloud native filesystem that can store and version all of your sequencing files.
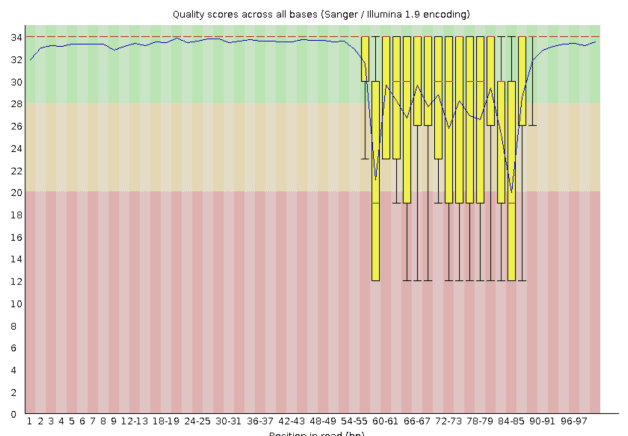
A biologist can then easily access, QC, organize, and explore their sequencing data. Scientists do not need anyone else to do this for them after it has uploaded.
Identify Enriched Library Members
Pooled library screens allow for hundreds, or even thousands, of candidate genes to be screened simultaneously. A bulk measurement is then taken from the pooled sequencing product, and computationally untangled to reveal which candidates are enriched or depleted in the treatment group.
Here a bioinformatician can use the Latch SDK to create production-grade QC and barcode counting workflows to process library members from raw sequencing files. They can easily iterate on the parameters and errors with biologists with full control over code.
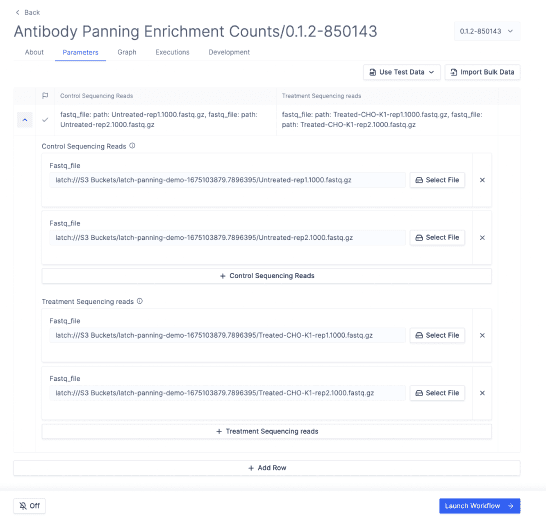
Here as a biologist, thank your bioinformatician. You now have a simple interface to analyze your enrichment data yourself.
Compare Library Performance across Conditions
When working with large-scale library screens under different selection conditions, it can be challenging to choose a “winner” given experimental variability. Statistical analysis is used to guide further investigation by narrowing in on lead candidates.
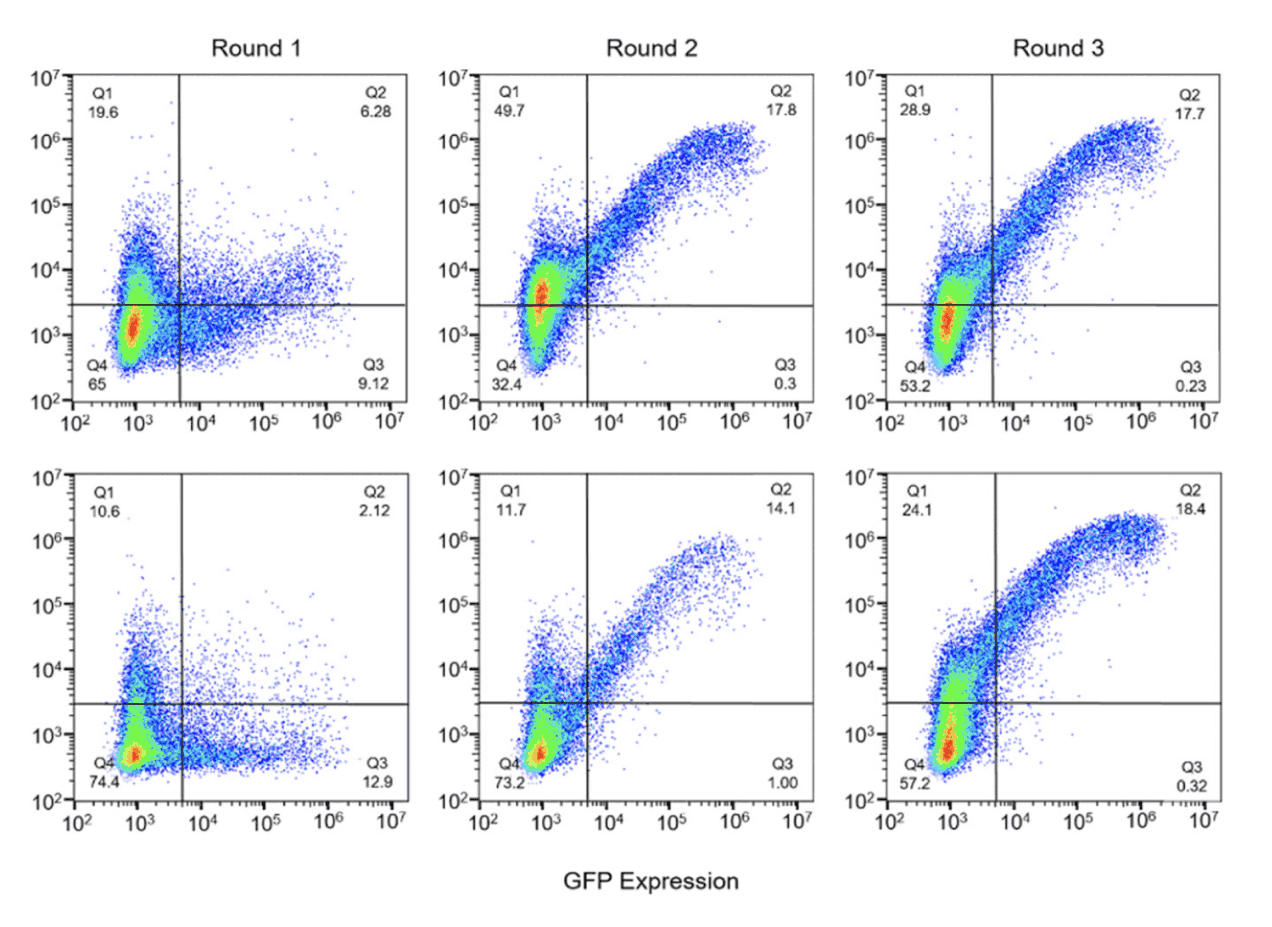
A bioinformatician can use Latch Pods to write flexible, custom jupyter or Rstudio notebooks to plot enrichment counts. Build an application to compile your results and expose it directly to your scientists without worrying about hosting or infrastructure.
The biologist can then easily interact with plots, allowing them to change filters and parameters on custom-built applications. For example, an application may display top performing library members from multiple enrichment experiments.
Organize Enrichment Data for Longitudinal Analysis
When evaluating libraries of candidate therapeutics against numerous conditions, the complexity of the associated sequencing data can quickly become unwieldy. It is critical to clearly register your past experimental data in an organized manner, to enable searching of previous results, contextualization of new data, and synthesis of new conclusions.
Latch Registry is like Notion or Google Sheets but designed for sample sheets. Registry lets you impose structure and organization on files, associating experimental inputs and outputs with metadata.
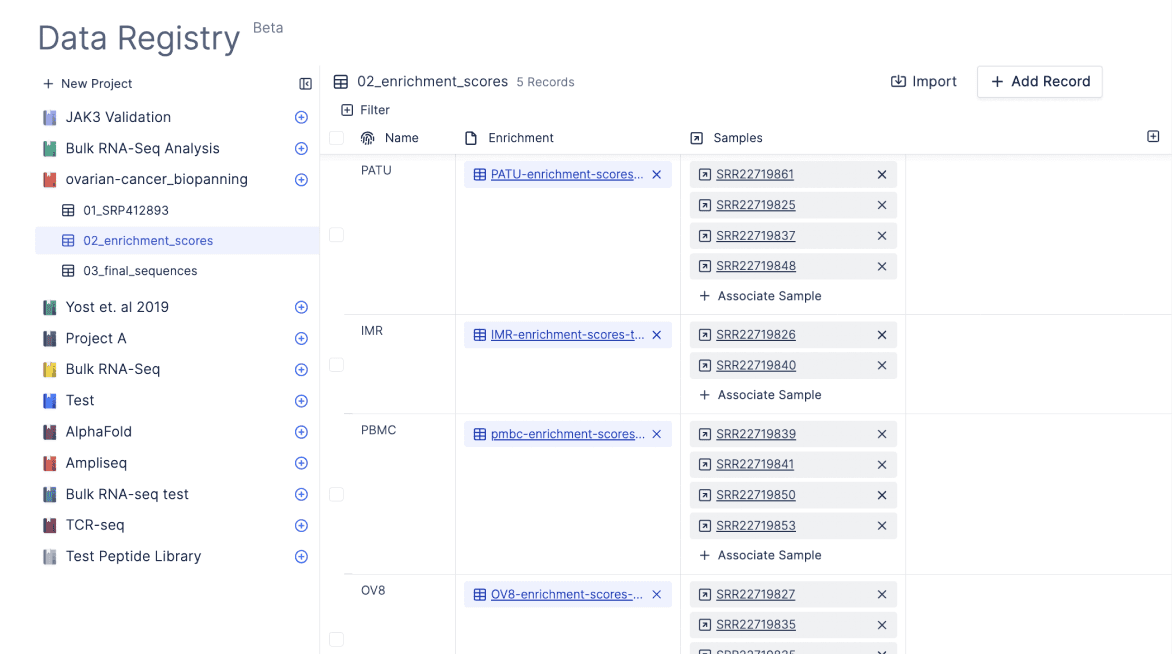
A bioinformatician team can build type-validated tables in Registry The bioinformatics are able to enforce accurate data entry, easily clean tables in code, and ingest them into bioinformatics workflows for batch processing.
The biologist can then go in and fill in the samplesheet metadata in a single place. They are easily able to view past experiments in a table that displays both experimental metadata and files in single location.
Previous
← Protein Engineering